

تالاسمی یک اختلالات خونی ارثی است که از طریق ژن از والدین به کودکان ارث می رسد. تالاسمی باعث می شوند بدن به سلول های قرمز و هموگلوبین سالم و کمتری دسترسی داشته باشد. هموگلوبین پروتئینی غنی از آهن در سلول های قرمز خون است. این پروتئین قادر است اکسیژن را به تمام نقاط بدن برساند. هموگلوبین همچنین دی اکسید کربن بدن که از طریق بازدم ریه ها تولید می شود را حمل می کند.
انواع تالاسمی
انواع مختلف تالاسمی وجود دارد که بستگی به شدت اختلال دارد. این موارد عبارتند از:
تالاسمی آلفا
- مرحله حامل خاموش
- تالاسمی آلفا مینور (حامل صفت تالاسمی)
- بیماری هموگلوبین H
- تالاسمی آلفا ماژور (هیدروپس فتالیس)
تالاسمی بتا
- تالاسمی بتا مینور (حامل صفت تالاسمی)
- تالاسمی بتا ماژور (آنمی کولی یا بتا صفر (SS0) تالاسمی)
- تالاسمی بتا پلاس (SS +)
چه عواملی باعث ایجاد تالاسمی می شوند؟
بدن شما سه نوع سلول خونی دارند که عبارتند از:
- سلول های قرمز خون
- سلول های سفید خون
- پلاکت ها
سلول های قرمز خون حاوی هموگلوبین، پروتئینی غنی از آهن هستند که اکسیژن را از ریه ها به تمام قسمت های بدن می رسانند. هموگلوبین دی اکسید کربن را نیز از بدن خارج می کند. هموگلوبین دارای دو نوع زنجیره ی پروتئین به نام گلوبین آلفا و گلوبین بتا می باشد. اگر بدن شما به اندازه کافی از این زنجیره پروتئین نداشته باشد و یا میزان آنها غیر طبیعی باشد، سلول های قرمز خون نمی توانند به درستی اکسیژن را در بدن حمل کنند. در صورت عدم وجود اکسیژن کافی نیز سیستم های بدن به خوبی کار نخواهد کرد. ژن ها، چگونگی تولید زنجیره های پروتئینی هموگلوبین را در بدن تحت کنترل دارند. هنگامی که این ژن ها از بین بروند یا تغییری در آن ها ایجاد شود، تالاسمی رخ می دهد.
تالاسمی اختلالی است که به ارث برده می شود. افرادی که ژن معیوب هموگلوبین را از یکی از والدین به ارث برده، و ژن دیگری که از والد دیگر خود دریافت می کند معمولی و طبیعی است، تنها ناقل هستند. حاملان ناقل، اغلب هیچ نشانه ای از بیماری کم خونی خفیف ندارند. با این حال، آنها می توانند ژن معیوب را به فرزندان خود منتقل کنند. افرادی که تالاسمی شدید دارند ژن معیوب را از هر دو والدین خود به ارث برده اند.
تالاسمی آلفا
به طور کلی هر فرد باید ۴ ژن (از هر والد دو ژن) از زنجیره های پروتئینی گلوبین آلفا به ارث ببرد. اگر یک یا چند ژن از بین برود، تالاسمی آلفا رخ می دهد. این به این معنی است که بدن شما به اندازه کافی پروتئین گلوبین آلفا ندارد.
- اگر تنها یک ژن از بین رفته باشد و یا ناقص باشد و ۳ ژن دیگر طبیعی باشد، فرد ناقل است. این به این معنی است که نشانه ای از بیماری در فرد وجود ندارد.
- اگر دو ژن از بین رفته و یا ناقص باشد و دو ژن دیگر در حالت طبیعی دریافت شده باشد، فرد دچار تالاسمی آلفا مینور شده. در این شرایط ممکن است فرد کم خونی خفیف را تجربه کند.
- اگر سه ژن از بین رفته و یا ناقص باشد و تنها یک ژن طبیعی وجود داشته باشد، فرد به احتمال زیاد بیماری هموگلوبین H دارد. این شکل از تالاسمی باعث آنمی متوسط تا شدید می شود.
- به ندرت پیش می آید که یک کودک هر ۴ ژن دریافتی اش از والدین ناقص و یا از بین رفته باشد در این شرایط فتالیس آلفا ماژور یا هیدروپس ایجاد می شود. نوزادانی که هیدروپس فتالیس دارند معمولا قبل از تولد یا مدتی پس از آن می میرند.
نمونه ای از یک الگوی وراثتی برای تالاسمی آلفا
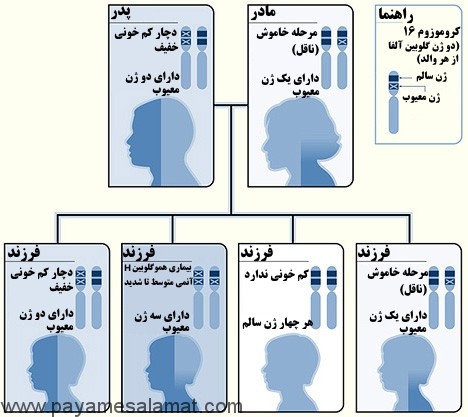
تصویر بالا یک مثال از چگونگی به ارث بردن تالاسمی آلفا را نشان می دهد. ژن گلوبین آلفا روی کروموزوم ۱۶ قرار دارد. کودک چهار ژن گلوبین آلفا (از هر والد دو ژن) دریافت می کند. در در این مثال، پدر دو ژن گلوبین آلفا ناقص و مادر یک ژن گلوبین آلفا ناقص دارد. در این شرایط هر کودک در مورد ابتلا به ۴ وضعیت زیر ۲۵ درصد شانس دارد:
- به ارث بردن دو ژن معیوب و دو ژن نرمال (تالاسمی مینور)
- به ارث بردن سه ژن معیوب و یک ژن نرمال (بیماری هموگلوبین H)
- به ارث بردن چهار ژن نرمال (بدون کم خونی)
- به ارث بردن یک ژن معیوب و سه ژن نرمال (مرحله حامل خاموش)
تالاسمی بتا
هر فرد باید دو ژن بتا (از هر والد یک ژن) از زنجیره های پروتئینی گلوبین بتا به ارث ببرد. اگر یکی یا هر دو این ژن ها تغییر یافته باشند، فرد به تالاسمی بتا مبتلا می شود. این به این معنی است که بدن شما به اندازه کافی پروتئین گلوبین بتا ندارد.
- اگر تنها یک ژن معیوب وجود داشته باشد، فرد ناقل است. این بیماری به نام تالاسمی بتا یا تالاسمی مینور بتا نیز شناخته می شود و باعث کم خونی خفیف خواهد شد.
- اگر هر دو ژن تغییر یافته باشد، فرد تالاسمی بتا ماژور (آنمی کولی) دارد. فرم متوسط از این اختلال باعث کم خونی متوسط و شکل اصلی آن باعث کم خونی شدید می شود.
نمونه ای از یک الگوی وراثتی برای تالاسمی بتا
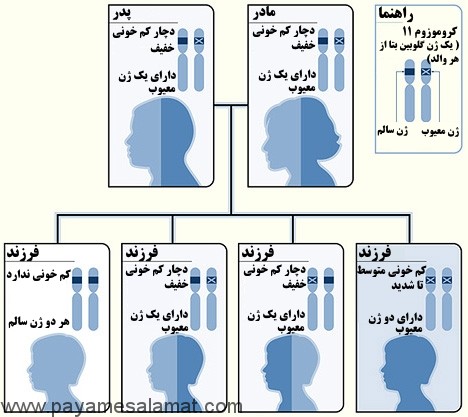
تصویر بالا یک مثال از چگونگی به ارث بردن تالاسمی بتا را نشان می دهد. ژن گلوبین بتا بر روی کروموزوم ۱۱ قرار دارد. کودک دو ژن گلوبین بتا (از هر والد یک ژن) دریافت می کند. در این مثال، هر یک از والدین یک ژن گلوبین بتا تغییر یافته دارند. در این شرایط هر کودک می تواند موراد زیر را تجربه کند:
- ۲۵ درصد شانس به ارث بردن دو ژن نرمال (بدون کم خونی)
- ۵۰ درصد شانس به ارث بردن یک ژن تغییر یافته و یک ژن طبیعی (تالاسمی بتا)
- ۲۵ درصد شانس به ارث بردن دو ژن تغییر یافته (تالاسمی بتا ماژور)
چه کسانی در خطر ابتلا قرار دارند؟
سابقه خانوادگی و اصل و نسب دو عوامل خطر برای ابتلا به این بیماری هستند.
سابقه خانوادگی
تالاسمی به معنی به ارث بردن ژن معیوب از پدر و یا مادر و یا هر دو آن ها است. پس اگر پدر و مادر ژن هموگلوبین معیوب و یا تغییر یافته داشته باشند، ممکن است کودکشان به تالاسمی مبتلا شود.
تبار
تالاسمی اغلب در میان ایتالیایی ها، یونانی ها، مردم خاورمیانه، جنوب آسیا و آفریقایی تبارها رخ می دهد.
علائم و نشانه تالاسمی چیست؟
فقدان اکسیژن در جریان خون باعث می شود که علائم و نشانه های تالاسمی خود را نشان دهند. اما شدت علائم بستگی به شدت اختلال دارد.
بدون نشانه
حاملان خاموش به طور کلی هیچ علائم و نشانه ای از این اختلال ندارند. در این مرحله، کمبود پروتئین گلوبین آلفا وجود دارد اما هموگلوبین به طور معمول در بدن فعال است.
کم خونی خفیف
افرادی که تالاسمی آلفا و یا بتا دارند، می توانند کم خونی خفیف نیز داشته باشند. کم خونی خفیف می تواند احساس خستگی را به بدن منتقل کند. کم خونی خفیف ناشی از تالاسمی آلفا ممکن است با کم خونی فقر آهن اشتباه گرفته شود.
دیگر علائم و نشانه های کم خونی خفیف و متوسط
افرادی که تالاسمی متوسط بتا دارند ممکن است سایر مشکلات بهداشتی، از قبیل موراد زیر را تجربه کنند:
- تاخیر در بلوغ و رشد: کم خونی می تواند سرعت رشد کودک را تحت تاثیر قرار دهد.
- مشکلات استخوان:تالاسمی ممکن است باعث شود مغز استخوان پهن شود. مغز استخوان ماده اسفنجی داخل استخوان است که سلول های خونی تولید می کند. هنگامی که مغز استخوان باز شود، استخوان از حد نرمال گسترده تر می شود. به همین دلیل ممکن است شکننده تر شود.
- بزرگ شدن طحال: طحال ارگانی در بدن است که در مبارزه با عفونت ها و حذف مواد ناخواسته، به بدن کمک می کند. هنگامی که یک فرد به این بیماری مبتلا می شود، طحال به سختی فعالیت می کند در نتیجه، بزرگتر از حد معمول می شود. این باعث می شود کم خونی بدتر شود. اگر طحال بیش از حد بزرگ شود، باید آن را برداشت.
علائم کم خونی شدید
افرادی که هموگلوبین H یا بیماری بتا ماژور (آنمی کولی) دارند به تالاسمی شدید مبتلا شده اند. علائم و نشانه ها در این بیماری معمولا در ۲ سال اول زندگی خود را نشان می دهند. این افراد ممکن است کم خونی شدید و سایر مشکلات، از جمله موارد زیر را تجربه کنند:
- ظاهر رنگ پریده
- کاهش اشتها
- ادرار تیره (نشانه آن است که سلول های قرمز خون در حال شکستن هستند)
- تاخیر در بلوغ و رشد
- یرقان (زرد شدن پوست و سفیدی چشم)
- بزرگی طحال، کبد و یا قلب
- مشکلات استخوان (به خصوص استخوان های صورت)
عوارض تالاسمی
امروزه درمان های زیادی وجود دارد که می تواند به افراد مبتلا به کم خونی متوسط و شدید کمک کنند تا عمر طولانی تری داشته باشند. اما در کنار این درمان ها انتظار می رود فرد بیمار عوارض زیر را تجربه کند.
قلب و کبد
انتقال خون منظم یک درمان استاندارد برای تالاسمی است. انتقال خون باعث می شود میزان آهن در بدن افزایش یافته و به اصطلاح بار آهن بیشتر شود. این عارضه می تواند به اندام ها و بافت ها، به خصوص قلب و کبد آسیب برساند. بیماری های قلبی ناشی از اضافه بار آهن علت اصلی مرگ در افرادی مبتلا به تالاسمی است. نارسایی قلبی، آریتمی (ضربان نامنظم قلب ) و حمله قلبی از جمله شایع ترین بیماری های قلبی برای این افراد است.
عفونت
در میان افرادی که کم خونی (تالاسمی) دارند، عفونت ها، علت اصلی بیماری و دومین علت شایع مرگ و میر است. افرادی که طحال خود را برداشته اند در معرض خطر بالاتری هستند زیرا آنها دیگر ارگان مبارزه کننده با عفونت را ندارند.
پوکی استخوان
بسیاری از افراد مبتلا به تالاسمی، مشکلات استخوان، استئوپروز دارند. این بیماری است که در آن استخوان های ضعیف و شکننده می شوند.
درمان تالاسمی
درمان این بیماری به نوع و شدت اختلال بستگی دارد. افراد حامل یا کسانی که علائمی ندارد و یا دارای علائم خفیف تالاسمی آلفا و بتا هستند، به احتمال زیاد نیاز به درمان ندارند و یا اگر درمان لازم باشد، نوع ساده و خفیف آن استفاده می شود. پزشکان از سه درمان استاندارد برای انواع متوسط و شدید تالاسمی، استفاده می کنند. این درمان ها شامل انتقال خون، درمان کی لیت آهن و مکمل های اسید فولیک است. درمان های دیگر در حال آزمایش هستند و کمتر از آن ها استفاده می شود.
انتقال خون
تزریق سلول های قرمز خون درمان اصلی برای افراد مبتلا به کم خونی متوسط یا شدید می باشد. این درمان، سلول های قرمز خون سالم با هموگلوبین طبیعی به بدن می رساند. در طول انتقال خون، سوزنی به یکی از وریدهای اصلی بدن وارد شده و از طریق آن خون سالم به بدن تزریق می شود. این روش معمولا ۱ تا ۴ ساعت طول می کشد.
سلول های قرمز خون فقط ۱۲۰ روز زنده هستند. بنابراین، برای حفظ منبع سالمی از سلول های قرمز خون، نیاز است انتقال خون مکررا انجام شود. البته اگر فردی به بیماری هموگلوبین H یا تالاسمی بتا مبتلا بود ممکن است در مناسب های مختلف، مثل زمانی که یک عفونت یا بیماری های دیگر وارد بدن شده باشد و یا وقتی کم خونی به اندازه کافی شدید شده و علت خستگی شود، نیاز به تزریق خون باشد.
اگر فردی آنمی کولی داشت، به احتمال زیاد نیاز به انتقال خون منظم (هر ۲ تا ۴ هفته یک بار) دارد. این انتقال کمک خواهد کرد که هموگلوبین و سطح سلول های قرمز خون نرمال بماند. تزریق خون به فرد بیمار احساس بهتری داده و موجب می شود از فعالیت های روزانه لذت بیشتری ببرد. این درمان نجات بخش است، اما در عین گران بودن، خطر انتقال عفونت و ویروس ها (برای مثال، هپاتیت) را به همراه دارد. با این حال، احتمال این خطرات به دلیل غربالگری دقیق خون، قبل از انتقال آن، بسیار کم است.
درمان کی لیت آهن
هموگلوبین در گلبول های قرمز خون پروتئینی غنی از آهن هستند. بنابراین، انتقال خون منظم می تواند به تجمع آهن در خون منجر شود. این وضعیت اضافه بار آهن نامیده می شود و ممکن است آسیب هایی برای کبد، قلب، و دیگر قسمت های بدن به همراه داشته باشد. برای جلوگیری از این آسیب، پزشکان از درمان کی لیت آهن، آهن زیادی بدن، استفاده می کنند. دو دارو برای درمان کی لیت آهن استفاده می شود.
دسفرال: یک داروی مایع است که معمولا با یک پمپ قابل حمل کوچک و به آرامی زیر پوست استفاده می شود. این روش درمانی هم طول می کشد و می تواند درد خفیفی داشته باشد و عوارض جانبی آن شامل مشکلات بینایی و شنوایی می باشد.
Deferasirox: قرصی است که باید روزانه مصرف شود. عوارض این دارو جانبی شامل سردرد، تهوع استفراغ، اسهال، درد مفاصل و خستگی است.
مکمل اسید فولیک
فولیک اسید جز ویتامین های گروه B است که کمک می کند تا سلول های قرمز خون سالم ساخته شود. معمولا پزشکان مکمل اسید فولیک را همراه با درمان تزریق خون و یا درمان کی لیت آهن توصیه می کنند.
درمان های دیگر
درمان های دیگر برای این بیماری در حال آزمایش هستند و از آن ها به ندرت استفاده می شود. این درمان عبارتند از:
پیوند سلول های بنیادی خون و مغز استخوان
در این روش، سلول های بنیادی مغز استخوان در افراد بیمار با سلول های بنیادی افراد سالم جایگزین می شود. سلول های بنیادی سلول هایی هستند که در داخل مغز استخوان قرار دارند. مغز استخوان وظیفه تولید سلول های قرمز خون و انواع دیگر سلول های خونی را به عهده دارد.
سایر درمان های در حال تحقیق
محققان در حال کار بر روی پیدا کردن درمان های جدید برای تالاسمی هستند. به عنوان مثال، ممکن است روزی قادر شوند ژن هموگلوبین نرمال را به سلول های بنیادی در مغز استخوان وارد کنند. محققان همچنین در حال مطالعه بر روی هموگلوبین جنینی پس از تولد می باشند. این نوع از هموگلوبین در جنین و نوزادان پیدا شده است و بعد از تولد، به هموگلوبین بزرگسالان تغییر می کند. ساخت هموگلوبین جنینی بیشتر ممکن است برای فقدان هموگلوبین بالغین مورد استفاده قرار گیرد.